





Mukowiscydoza
CO TO JEST MUKOWISCYDOZA ?
Mukowiscydoza (Mucoviscidosis, cystic fibrosis, torbielowate zwłóknienie trzustki) jest to wielonarządowa choroba dzieci i dorosłych, uwarunkowana genetycznie. W klasycznej (pełnoobjawowej) postaci objawia się skłonnością do zapalenia oskrzeli i płuc, niewydolnością części zewnątrzwydzielniczej trzustki, niepłodnością mężczyzn oraz podwyższonym stężeniem chlorków w pocie.
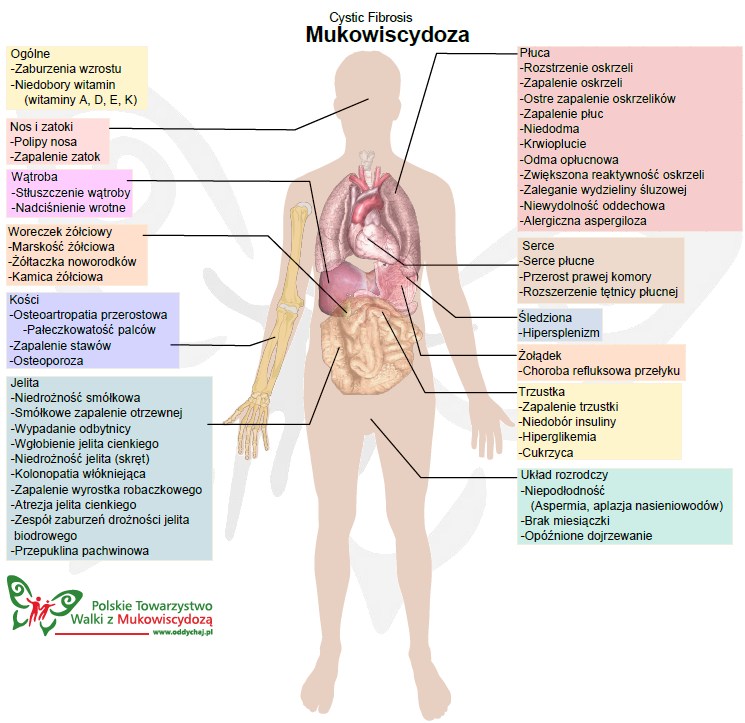
Mukowiscydoza jest jednym z ważniejszych problemów pediatrycznych, stanowiąc częstą przyczynę ciężkich i przewlekłych zmian w układzie oddechowym, niewydolności zewnątrzwydzielniczej trzustki, polipowatości błony śluzowej nosa, zapaleń zatok obocznych nosa, wypadania błony śluzowej odbytu, zaburzeń odżywiania, czasami marskości wątroby i innych form dysfunkcji tego narządu.
Zwiększona lepkość wydzieliny oskrzelowej w mukowiscydozie jest wynikiem wytwarzania nieprawidłowych mukoprotein, niedostatecznego uwodnienia oraz uwalniania znacznych ilości DNA z neutrofilów rozpadających się w oskrzelach w odpowiedzi na przewlekłe zakażenie. Zawartość DNA w ropnej treści oskrzelowej u chorych na mukowiscydozę przekracza kilkakrotnie stężenia występujące w innych chorobach oskrzeli.
Częstość występowania mukowiscydozy w Polsce oceniana jest na 1:2300żywo urodzonych dzieci. Chorobą tą dotknięte są zarówno noworodki, niemowlęta, dzieci starsze, jak i dorośli. Długość przeżycia, jak i jakość życia pacjentów w dużej mierze zależą od wczesnego rozpoznania i prawidłowego leczenia. Mimo znacznego postępu konwencjonalnych metod terapii, średnia długość życia chorych wynosi 30 lat.
Przy częstości urodzeń 400.000, w Polsce rocznie rodzi się około 200 dzieci dotkniętych tą chorobą. Około dwóch milionów Polaków, w równym stopniu mężczyzn i kobiet, jest nosicielami zmutowanego genu CFTR.
Dane z kwietnia 2007 roku mówią o 1134 osobach chorych na mukowiscydozę w Polsce.
OBJAWY
1. Układ oddechowy
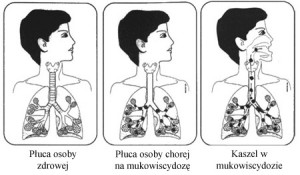
Pierwsze objawy kliniczne ze strony układu oddechowego występują najczęściej w wieku niemowlęcym. Zastój śluzu inicjuje zapalenie oskrzelików, którego początek wiązany jest z banalną infekcją wirusową.Męczący, uporczywy, nieproduktywny kaszel, zadyszka lub duszność traktowane są jako objaw infekcji wirusowej. Gęsty śluz czopuje oskrzeliki powodując rozdęcie i odcinkową niedodmę płuc.U większości dzieci dochodzi do zakażenia gronkowcem złocistym, pałeczką grypy, a często również pałeczką ropy błękitnej. W dalszym przebiegu choroby dominuje ropne zapalenie oskrzeli i płuc z okresowymi remisjami i zaostrzeniami. Postępujące zapalenie obejmuje niedodmowe obszary tkanki płucnej. Obraz morfologiczny nie jest jednolity. W różnych obszarach płuc równocześnie może występować rozdęcie, rozedma, niedodma, zapalenie płuc, włóknienie płuc, rozstrzenie oskrzeli, pęcherze rozedmowe, torbiele. Wraz z postępem choroby i przewlekłym niedotlenieniem rozwija się serce płucne.Już we wczesnym okresie choroby mogą utworzyć się palce pałeczkowate – powodowane niedotlenieniem peryferyjnych części ciała. U dzieci starszych i u dorosłych mogą występować poważne powikłania, jak: krwawienia płucne, odma opłucnej oraz zniekształcenie klatki piersiowej z pogłębieniem przednio – tylnego wymiaru.
W górnych drogach oddechowych rozwija się ropne zapalenie zatok bocznych nosa, którego podłożem jest zaleganie śluzu. U około 20% chorych przewlekłe ropienie jest przyczyną polipów nosa.Ropne zapalenie zatok występuje u prawie wszystkich chorych dorosłych. W badaniu rentgenowskim jednym z pierwszych uchwytnych objawów jest nadmierna przejrzystość pól płucnych odpowiadająca rozdęciu płuc. Nadmierna powietrzność jest stałym objawem rentgenowskim niezależnie od wieku chorego. Początkowo jest to rozdęcie płuc, a z postępem choroby staje się wynikiem rozedmy. Na tle nadmiernej powietrzności płuc występują zacienienia ogniskowe, segmentowe lub płatowe, odcinkowe lub rozsiane śródmiąższowe włóknienie płuc, cechy rozstrzeni oskrzeli i pęcherze rozedmowe. Węzły chłonne wnęk zwykle są powiększone jako wynik przewlekłego zapalenia.
2. Układ pokarmowy
Ponad 80% dzieci chorych na mukowiscydozę cechuje upośledzenie rozwoju somatycznego związane z niewydolnością zewnątrzwydzielniczą trzustki. Chorych charakteryzuje zwykle oddawanie tłuszczowatych stolców z niestrawionymi resztkami pokarmowymi, zaniżenie wskaźników rozwoju fizycznego zarówno masy ciała, jak i wysokości. Dość częstym objawem u dzieci do 3 roku życia jest wypadanie śluzówki odbytu.
Najwcześniejszą postacią kliniczną mukowiscydozy może być niedrożność smółkowa spowodowana nagromadzeniem zagęszczonej smółki w jelitach.W badaniu radiologicznym jamy brzusznej poza charakterystycznym objawem rozdęcia pętli jelitowych bez poziomów płynu (duża lepkość smółki) mogą być widoczne zwapnienia, gdy powikłanie (perforacja jelita i zapalenie otrzewnej) nastąpiło w życiu płodowym.
Niewydolność zewnątrzwydzielnicza trzustki. Do niewydolności zewnątrzwydzielniczej trzustki w przebiegu mukowiscydozy dochodzi w następstwie gromadzenia się w obrębie układu przewodów wyprowadzających sok trzustkowy gęstego, lepkiego śluzu. W wyniku tej blokady rozpoczyna się autoliza (samoniszczenie) komórek gruczołowych i zanik miąższu trzustki (on to właśnie produkuje prawidłowy sok trzustkowy) z zastępowaniem tkanki gruczołowej przez tkankę włóknistą co doprowadza do niewydolności narządu. Stopień upośledzenia zewnątrzwydzielniczej czynności trzustki jest różny i przyjmuje się, że u około 10 – 20% chorych może nie doprowadzać do klinicznego ujawnienia się niedoborów odżywienia.Jednak wiadomo również, że nieprawidłowo, lub niewystarczająco uzupełnianie niedobory enzymatyczne występujące w przebiegu choroby doprowadzać mogą z wiekiem do ujawnienia się, utajonego wcześniej, uszkodzenia trzustki. Tak więc raz jeszcze należy podkreślić, że niewydolność zewnatrzwydzielnicza trzustki może występować już od chwili narodzin dziecka z mukowiscydozą (opisywano także zmiany u płodów) i jest nieodłączną składową procesu chorobowego.
Niedrożność smółkowa. Z wiekiem chorych zwiększa się częstość występowania ekwiwalentów niedrożności smółkowej, czyli zespołu DIOS (distal intestinal obstruction syndrom), objawiającego się bólami brzucha, miękkim guzem w prawym podbrzuszu, radiologicznie cechami zalegania mas kałowych.W mukowiscydozie częściej niż w ogólnej populacji stwierdza się występowanie refluksu żołądkowo – przełykowego.Do „efektów ubocznych” choroby należą także przedłużająca się żółtaczka noworodkowa, marskość wątroby, kamica żółciowa.Niezależnie od objawów upośledzenia czynności zewnątrzwydzielniczej trzustki dochodzi czasami do zaburzeń endokrynnych pod postacią cukrzycy. Dotyczy to na ogół chorych powyżej 10 roku życia.
Niedożywienie. Niedożywienie w mukowisdcydozie jest uwarunkowane niewydolnością trzustki i złym wchłanianiem, brakiem łaknienia, częstymi infekcjami przebiegającymi z gorączką, zwiększonym wydatkiem energetycznym, zwiększonym rozkładem białek ustrojowych w wyniku zakażeń układu oddechowego itp. Prowadzi to do pogorszenia wydolności układu oddechowego i osłabienia odporności. Wcześniej dochodzi do stanu zmęczenia i osłabienia mięśni oddechowych i mięśni szkieletowych, co oznacza niższą tolerancję wysiłku fizycznego – gorszą wydolność przy ćwiczeniach rehabilitacji oddechowej. Utrata masy ciała rzędu 10 – 20% może mieć niekorzystny wpływ na wszystkie funkcje organizmu. Dochodzi do upośledzenia pracy mięśni, osłabienia układu oddechowego, układu odpornościowego i mechanizmów termoregulacji, obniża się odporność na zakażenia. Może też dochodzić do pogorszenia wydolności układu krążenia i przewodu pokarmowego.
3. Układ rozrodczy
Mężczyźni. U mężczyzn chorych na mukowiscydozę w badaniach mikroskopowych jąder stwierdza się niewielkie zmiany. Przede wszystkim jest to zmniejszenie ilości komórek Sertoliego ułatwiających dojrzewanie plemników oraz obecność obszarów powstawania nieprawidłowych plemników. Całkowita liczba dojrzałych plemników jest obniżona i stwierdza się zwiększony odsetek plemników nieprawidłowych. Powyższe zaburzenia powodują, że 97 – 98% mężczyzn chorych na mukowiscydozę jest bezpłodnych.
Kobiety. U kobiet chorych na mukowiscydozę stwierdza się zwiększone wytwarzanie nadmiernie gęstego i lepkiego śluzu w szyjce macicy. Zawiera on mniej wody i elektrolitów. Nie występuje zjawisko „upłynnienia” śluzu obserwowane u zdrowych kobiet w czasie owulacji (jajeczkowania). Zaleganie gęstego śluzu jest najprawdopodobniej przyczyną dość częstego powstawania polipów śródszyjkowych. Jednocześnie lepki śluz utrudniając przechodzenie plemników zmniejsza szansę zapłodnienia. Jeżeli już dojdzie do zapłodnienia, to utrudnione jest zagnieżdżenie się zapłodnionej komórki jajowej w ścianie macicy. To wszystko powoduje, że szansę na zajście w ciążę u kobiet chorych na mukowiscydozę są około czterokrotnie mniejsze niż w całej populacji.
Każda przewlekła choroba, w tym również mukowiscydoza wpływa na czas wystąpienia pierwszej miesiączki. U dziewcząt chorych na mukowiscydozę dojrzewanie jest opóźnione względem zdrowych koleżanek o około 2 – 3 lata.Im bardziej nasilone są zmiany płucne, zaburzenia ze strony układu pokarmowego jak i niedobory odżywienia, tym później pojawia się pierwsza miesiączka. Ciężki przebieg choroby może spowodować, że miesiączka nigdy nie wystąpi (tzw. pierwotny brak miesiączki). Również nasilenie choroby u kobiety wcześniej miesiączkujacej może przyczynić się do zatrzymania miesiączki (tzw. wtórny brak miesiączki) lub występowania cyklów bezowulacyjnych, a więc takich w których nie dochodzi do uwalniania komórki jajowej z jajnika i niemożliwe jest zapłodnienie.
LECZENIE MUKOWISCYDOZY
1. Układ oddechowy
Ewakuacja wydzieliny oskrzelowej. Do podstawowych metod ewakuacji wydzieliny oskrzelowej należą zabiegi fizjoterapeutyczne. Klasyczne metody fizjoterapii obejmują konwencjonalny drenaż ułożeniowy z oklepywaniem, wibracją klatki piersiowej oraz drenaż z aktywnymi, przedłużonymi, forsownymi wydechami wspomaganymi uciskiem na ścianę klatki piersiowej. Innym sposobem „pozbycia się” wydzieliny z układy oddechowego jest oddychanie z dodatnim ciśnieniem wydechowym.Podwyższony opór wydechowy zapobiega przedwczesnemu zapadaniu się oskrzeli ułatwiając przemieszczanie wydzieliny z drobnych oskrzeli do tchawicy. W tym celu stosowana jest maska PEP (Positive Expiratory Pressure). Podobną zasadę ma Flutter, który stwarza przerywane, dodatnie ciśnienie wydechowe ułatwiając dodatkowo odrywanie się wydzieliny od ścian dróg oddechowych.
Leki mukolityczne. Najsilniej działającym lekiem mukolitycznym jest Dornaza Alfa (Pulmozyme) – lek ten jest otrzymywany na drodze inżynierii genetycznej (rekombinacja DNA). Działanie Dornazy polega na hydrolizie endogennego DNA uwalnianego z granulocytów rozpadających się w wyniku odpowiedzi zapalnej, powodując tym samym zmniejszenie lepkości wydzieliny oskrzelowej. Pulmozyme zmniejsza obturację oskrzeli i w długotrwałych obserwacjach ogranicza użycie antybiotyków.
Leki rozszerzające oskrzela. Charakterystyczną cechą przebiegu mukowiscydozy jest obturacja oskrzeli. Ocenia się, że około połowy chorych wykazuje dodatni wynik testu prowokacyjnego na histaminę, metacholinę lub zimne powietrze. Zastosowanie bronchodilatatorów u chorych mukowiscydozę nie daje jednoznacznie pozytywnego efektu, a według niektórych autorów nawet pogorszenie wskaźników czynności płuc. Bronchodilatatory mają uzasadnienie stosowania u starszych dzieci i u dorosłych, którzy wykazują dodatni test odwracalności obturacji. Leki sympatykomimetyczne obok działania rozszerzającego oskrzela wykazują mobilizujący transport śluzowo – rzęskowy.
Leczenie zakażenia bakteryjnego. Standardowym leczeniem zaostrzeń oskrzelowo – płucnych zwłaszcza wywołanych przez Pseudomonas aeruginosa jest podawanie dożylne dwu antybiotyków: aminoglikozydu (amikacyna, gentamycyna, tobramycyna, netilmycyna) z cefalosporyną trzeciej generacji (ceftazydym, cefsulodyna) lub półsyntetyczną penicyliną(azlocylina, piperacylina, karbenicylina, timentyna). Kombinacje tych antybiotyków są często skuteczne również w leczeniu zakażeń Staphylococcus aureus oraz Haemophilus influenzae. Do antybiotyków o dużej skuteczności w leczeniu zakażeń wymienionymi patogenami należy ciprofloksacyna, o podobnej skuteczności tak przy podaniu doustnym, jak i dożylnym oraz imipenem z cilastatyną.Chloramfenikol jak i kotrimoksazol są użyteczne w leczeniu niezwykle opornych na leczenie zakażeń Burkholderia (Pseudomonas) cepacia. Narastającym problemem jest zakażenie metycylinoopornymi szczepami gronkowca złocistego (MRSA) występujące nawet u kilkudziesięciu procent chorych na mukowiscydozę. Wysoce skutecznym antybiotykiem w leczeniu tego szczepu jest wankomycyna.
Leczenie chirurgiczne. Ostatecznym krokiem w leczeniu mukowiscydozy jest przeszczep płuc, polegający na usunięciu zmienionej chorobowo części płuc. Niestety taka operacja jest kosztowna.A jeszcze większym problemem niż koszt operacji jest brak dawcównarządów do przeszczepu. W ostatnich latach odnotowuje się wzrost liczby chorych umierających w oczekiwaniu na dawcę, odsetek ten sięga w niektórych ośrodkach 80%spośród wszystkich chorych zakwalifikowanych do przeszczepu
2. Układ pokarmowy
Enzymy trzustkowe. U chorych na mukowiscydozę dochodzi do ujemnego bilansu energetycznego, na skutek zarówno utraty energii, jak również zwiększonego zapotrzebowania na dodatkowa, wzmożoną pracę mięśni oddechowych i ich zwiększony metabolizm.Do ujemnego bilansu energetycznego prowadzi w tych przypadkach dodatkowo niedostateczne spożycie pokarmów, jako wynik obniżonego łaknienia.
Wykorzystanie wszystkich składników pokarmowych (białek, tłuszczów, węglowodanów) z pożywienia jest ograniczone z powodu niedostatecznego wydzielania enzymów trzustkowych w układzie pokarmowym, a przede wszystkim w trzustce. W następstwie tych zmian, związanych z upośledzonym trawieniem i niedostatecznym wchłanianiem wszystkich składników odżywczych, a także zbyt szybkim przechodzeniem treści pokarmowej przez przewód pokarmowy, towarzyszącymi bólami brzucha, wzdęciami i obfitymi stolcami tłuszczowymi („diabelskie koło” energetyczne), dochodzi do niedożywienia objawiającego się niedoborami masy i wysokości ciała, ogólnym osłabieniem, apatią i pogorszeniem funkcji płuc.
By zapewnić wyżej opisane zasady leczenia żywieniowo – enzymatycznego chorych na mukowiscydozę należy zdać sobie sprawę z tego, że zaburzenia wchłaniania występujące u około 80 – 90% chorych spowodowane są tzw. niewydolnością zewnątrzwydzielniczą trzustki i ona przede wszystkim powinna zostać uzupełniona.
Dieta wysokokaloryczna i wysokobiałkowa. Aby utrzymać prawidłową masę ciała chory powinien spożywać 120 – 140% normy zapotrzebowania energetycznego. Stały spadek masy ciała, niski wskaźnik BMI i niedobory składników stwierdzone w ocenie jadłospisów są wskazaniem do włączenia intensywnego dożywiania przez nocną sondę dożołądkową lub przez gastrostomię.Uzyskanie wysokiego spożycia energii jest możliwe tylko poprzez zwiększone spożycie tłuszczu w diecie. W diecie należy korzystać wyłącznie z tłustego mleka, tłustych serów, twarogów i jogurtów, do zup i sosów używać dodatku śmietany, do ziemniaków, ryżu i makaronów – dodawać masło, potrawy z mięsa i ryb przygotowywać z dodatkiem tłuszczu.
Do sałatek i surówek należy używać przede wszystkim tłuszczów roślinnych na surowo – oliwy z oliwek, oleju sojowego, słonecznikowego lub oleju z pestek winogron,które są przede wszystkim źródłem niezbędnych nienasyconych kwasów tłuszczowych (NNKT) – składników strukturalnych wszystkich błon komórkowych organizmu, o szczególnym znaczeniu m. in. dla funkcji komórek nabłonka układu oddechowego.
Należy unikać picia zwykłej wody lub napojów o znikomej wartości odżywczej (herbata z cukrem, napoje gazowane), zastępując je napojami mlecznymi (jogurty, koktajle mleczne, kakao).Posiłki powinny być niezbyt obfite, ale regularne i zawsze pełnowartościowe.
Witaminy. Mimo zalecanej rutynowej suplementacji wysokimi dawkami witamin rozpuszczalnych w tłuszczach (ADEK), okresowo powinno się oceniać ich stężenie w osoczu, co pozwoli dobrać dawkę najbardziej skuteczną, zabezpieczającą zapotrzebowanie organizmu. Zapotrzebowanie na witaminy rozpuszczalne w wodzie (witaminy z grupy B, wit. C) jest zgodne z normami dla osób zdrowych w tym samym wieku.
ROZPOZNANIE MUKOWISCYDOZY
1. Test potowy
Ilościowe oznaczanie elektrolitów w pocie jest badaniem weryfikującym rozpoznanie stawiane na podstawie objawów klinicznych. W celu uzyskania próbki potu do badań ilościowych stosuje się powszechnie metodę jontoforezy pilokarpinowej wprowadzoną w 1959 roku przez Gibsona i Cooka, a następnie zmodyfikowaną przez Shwachmana. Stężenia sodu i chloru powyżej 60 mmol/l uważane są za wartości patognomoniczne dla chorych na mukowiscydozę. Ten test jest powszechnie stosowany w diagnostyce. Zespoły chorobowe będące przyczyną podwyższonych elektrolitów w pocie poza mukowiscydozą to między innymi: niewydolność kory nadnerczy, dysplazja ektodermalna, ciężkie niedożywienie, nerkowopochodna moczówka prosta, niedoczynność tarczycy, niedoczynnośc przysadki, glikogenoza mukopolisacharydoza.
2.Test konduktometryczny Wescor.
Służy on do określania przewodnictwa elektrycznego potu. Jest alternatywną metodą dla testu klasycznego, zwłaszcza w ośrodkach nie dysponujących zapleczem laboratoryjnym. Wyniki nieprawidłowe wymagają weryfikacji klasyczną metodą testu potowego.
3. Pomiar przeznabłonkowej różnicy potencjałów w nosie.
U chorych na mukowiscydozę stwierdza się zwiększoną, bardziej ujemną, różnicę potencjałów nosa niż u zdrowych, związaną z zaburzeniami funkcji kanału chlorkowego. Pomiar wykonywany jest przez wprowadzenie elektrody (cewnik Foley’a) do nosa poniżej małżowiny dolnej. Elektroda referencyjna umieszczona jest na uprzednio poddanej punktowej abrazji skórze przedramienia. Wynik jest średnią z trzech pomiarów wykonanych w każdym przewodzie nosowym.
4.Diagnostyka molekularna.
Polega na wykryciu u chorego na mukowiscydozę dwóch mutacji genu CFTR na homologicznych chromosomach,co stanowi weryfikację rozpoznania klinicznego mukowiscydozy. Jest już jednak znanych ponad 600 mutacji, tak więc praktyczne zastosowanie w diagnostyce klinicznej obejmuje oznaczanie tylko kilku najczęściej występujących.
DZIEDZICZENIE MUKOWISCYDOZY
Jedną z najprostszych metod przedstawienia prawdopodobieństwa dziedziczenia cech jest metoda wynaleziona na początku XX wieku przez angielskiego genetyka, Reginalda Punetta. Za pomocą prostych kwadratów możliwe jest wyznaczenie prawdopodobieństwa, z jakim może wystąpić dany genotyp, a więc również dana choroba. Prawdopodobieństwo zachorowania na CF przedstawiają poniższe tabele.
Oznaczenia poszczególnych genotypów:
· Osoba zdrowa oznaczona jest genotypem (AA)
· Nosiciel (heterozygota) oznaczony jest (Aa)
· Osoba chora posiada dwie kopie zmutowanego genu. Jej genotyp oznaczono (aa)
Przypadek 1
Rodzice mają genotyp (Aa) i (Aa) (nosiciele – heterozygoty). W tym przypadku prawdopodobieństwo wynosi:
|
Tab. 1 |
A |
a |
|
A |
AA |
Aa |
|
a |
Aa |
aa |
25% ich potomstwa będzie zdrowe (AA)
50% ich potomstwa będzie nosicielem bez objawów klinicznych(Aa)
25% ich potomstwa będzie chore na CF (aa)
Przypadek 2.
Jeden rodzic jest chory na CF (aa), drugi jest zdrowy (AA). W tym przypadku prawdopodobieństwo wynosi:
|
Tab. 2 |
A |
A |
|
a |
Aa |
Aa |
|
a |
Aa |
Aa |
100% ich potomstwa będzie nosicielami bez objawów klinicznych (Aa)
Przypadek 3.
Jeden rodzic jest chory na CF (aa), drugi jest nosicielem (Aa). W tym przypadku prawdopodobieństwo wynosi:
|
Tab. 3 |
A |
a |
|
a |
Aa |
aa |
|
a |
Aa |
aa |
50% ich potomstwa będzie nosicielem bez objawów klinicznych (Aa)
50% ich potomstwa będzie chore na CF (aa)
HISTORIA MUKOWISCYDOZY
Mukowiscydoza kiedyś
Mukowiscydoza (zwłóknienie torbielowate trzustki, ang. cystic fibrosis – CF) jest to choroba, która dotyka ludzi od wielu tysięcy lat. Od czasu identyfikacji genu choroby istnieją możliwości określenia daty i miejsca pochodzenia CF. Hiszpański naukowiec Xavier Estivill z Instytutu Badań Raka w Barcelonie ocenił w 1993 roku, że wiek najczęstszej mutacji w obrębie genu CFTR – delta F508 wynosi przynajmniej 52000 lat!!!
CF rozprzestrzeniła się poprzez migrację ludów i kolonializm. W dawnych czasach, kiedy nie znano odpowiedniego postępowania leczniczego przeżycie okresu młodości chorych z CF graniczyło z cudem. Prawdopodobnie jednak w przypadku epidemii cholery chorzy na CF mieli większą szansę przeżycia niż zdrowi ludzie. Sugeruje to współcześnie przedstawione doświadczenie przeprowadzone na myszach. Wykazano, że mysz laboratoryjna nosicielka genu CF nie ginie od biegunki typowej dla cholery, gdyż prawdopodobnie upośledzony jest transport jonów chlorkowych, co obniża „ciężkość” przebiegu cholery przez ograniczenie utraty wody z jelit. Przenosząc tę hipotezę na ludzi nosiciele genu CF – heterozygoci przeżywali cholerę „łatwiej”, nie – nosiciele genu umierali. Tym mechanizmem tłumaczy się również częstsze występowanie nosicielstwa CF wśród rasy białej – „Europejczyków z pochodzenia”.
Prawdopodobnie za najwcześniejsze wiarygodne doniesienie medyczne dotyczące CF można uznać opis badania pośmiertnego 11 letniej dziewczynki, które wykonał w dniu 16 stycznia 1595 roku Peter Paaw profesor botaniki i anatomii w Leiden (Holandia). „Dziewczynkę podejrzewano, że jest zaczarowana. Chora była skrajnie wychudzona i wyczerpana trawiącą gorączką” – pisał profesor Paaw, uznając, że: „przyczyną zgonu była stwardniała i zwłókniała trzustka”. Jest to nie tylko pierwszy historycznie opis CF, ale wytłumaczenie zabobonów medyczną wiedzą XVI wieku.
Drugi przypadek opisu CF pochodzi również z Holandii. Gerardus Blasius, dyrektor szpitala w Amsterdamie, w książce p.t.: „Observationes Medicae Rariores” (Obserwacje niezwykłości medycznych) z 1677 roku, opisuje włóknienie trzustki u 9 – letniego chłopca zmarłego wśród objawów skrajnego wyniszczenia .
Trzeci opis pochodzi od niemieckiego lekarza Georga Segera, który w 1673 roku leczył w Toruniu około 3 letnią dziewczynkę z upośledzonym rozwojem fizycznym, przewlekle gorączkującą, cierpiącą na biegunkę i wymioty. W badaniu pośmiertnym stwierdzono, że jedyną zmianą chorobową była stwardniała trzustka.
W XIX wieku i wczesnych latach XX wieku niektóre doniesienia wiązały ze sobą występowanie biegunki tłuszczowej, powikłań niedrożności smółkowej i uszkodzeń trzustki. W 1905 roku Landsteiner, który także opisywał grupy krwi opublikował prawdopodobnie jako pierwszy współczesny opis niedrożności smółkowej u noworodków powiązanej ze zwłóknieniem torbielowatym trzustki. Nadejście XX wieku przyniosło pierwsze obserwacje, w których zaczęto wiązać ze sobą występowanie chorób płuc i biegunki tłuszczowej.
W 1919 roku Passini opisał dwoje dzieci zmarłych z powodu zapalenia płuc, u których wykazał współistnienie zmian torbielowatych trzustki. W 1928 i w 1936 roku Fanconi wybitny pediatra szwajcarski zwrócił uwagę na grupę chorych, u których w odróżnieniu od choroby trzewnej (celiakii) zaburzenia trawienia występowały od najwcześniejszego dzieciństwa i u których często stwierdzano zapalenia oskrzeli i rozstrzenie oskrzeli. Opisy choroby w tym okresie skupiały się na niewydolności trzustki i biegunce tłuszczowej, często odnoszono je do choroby trzewnej. Odnotowywano też współistnienie powikłań oskrzelowo płucnych. Przed 1938 rokiem kiedy choroba została poznana, umieralność przed 3 rokiem życia wynosiła 95%.
Mukowiscydoza teraz
Współczesna historia mukowiscydozy datuje się od 1938 roku. W tym roku Dorothy Anderson w użyła po raz pierwszy nazwy: „cystic fibrosis of the pancreas” (zwłóknienie torbielowate trzustki), wyodrębniła mukowiscydozę jako odrębną jednostkę nozologiczną. Jej zasługą jest pierwsza definicja CF jako choroby, której typowymi objawami są kaszel, luźne stolce, opóźnienie rozwoju. Anderson konkludowała: „istnieje bliski związek pomiędzy biegunką tłuszczową, zwłóknieniem torbielowatym trzustki i zapaleniami dróg oddechowych”.
W 1945(przed wykryciem zmian w pocie) Faber użył terminu mucoviscidosis. W przeciwieństwie do Anderson, która zaproponowała nazwę odnoszącą się do zmian w trzustce uważał on, że choroba dotyczy gruczołów zewnątrzwydzielniczych, a jej przyczyną jest stan nadmiernego zagęszczenia śluzu. Nazwa mucoviscidosis jest nadal szeroko używana i często preferowana w krajach nie anglojęzycznych.
W 1953 roku współpracownicy Dorothy Anderson: Paul di Sant’Agnese oraz Darlong, Perera i Shea dokonali znaczącego odkrycia stwierdzając nieprawidłowy skład elektrolitów w pocie u chorych na CF. Rzuciło to nowe spojrzenie na wczesne spostrzeżenie „słono smakujących pocałunków”. Powszechne zaakceptowanie faktu, że jednym z zaburzeń fizjologicznych w CF są nieprawidłowości w składzie elektrolitów zajęło kilka lat. Umożliwiło to opracowanie metody diagnostycznej przydatnej w rozpoznawaniu choroby.
Znaczące naukowo doniesienie o nieprawidłowościach w transporcie jonu chlorowego pojawiło się w 1968 roku, jednak dopiero we wczesnych latach 80 wyjaśnił je Quinton.Uznano to wówczas za jedno z najistotniejszych zaburzeń fizjologicznych w CF. Pomimo intensywnych poszukiwań, rozwoju nauki i pojawiania się nowych teorii dotyczących rozwoju choroby patofizjologia CF nie jest do końca wyjaśniona i stanowi wielkie wyzwanie dla świata nauki. Od czasu scharakteryzowania CF dokonano znaczącego postępu w zakresie leczenia tej choroby. Antybiotyki użyto po raz pierwszy w leczeniu chorych na CF w 1949 roku. Były to terramycyna, aureomycyna, chloromycetyna. Rola fizykoterapii klatki piersiowej w CF pozostawała niedoceniona do 1956 roku.
Kamieniem milowym zmian w postępowaniu terapeutycznym stały się przeprowadzone w Bostonie i Toronto badania wskazujące na wpływ zróżnicowanego postępowania żywieniowego na przebieg choroby. Opublikowali je w 1988 roku Corey. Oceniono w nich długość życia, rozwój fizyczny, częstość infekcji płucnych w 2 populacjach pacjentów o podobnej charakterystyce. Obie grupy obejmowały powyżej 500 chorych na CF. W Bostonie stosowano dietę niskotłuszczową i wysokowęglowodanową; w Toronto nie ograniczano podaży tłuszczów i podawano enzymy trzustkowe. W populacji z Bostonu średnia przeżycia 50% pacjentów wynosiła 21 lat, a w populacji z Toronto 30 lat. Zbliżenie lub wręcz ujednolicenie zaleceń związanych z szeroko pojmowaną terapią i opieką medyczną nad pacjentem z CF w USA i Kanadzie dało szybkie efekty.
Duże nadzieje w postępie terapii chorych na CF wiąże się z genoterapią. Nadal jednak jest to jedynie „metoda przyszłości”. Autorzy wcześniej publikowanych prac byli świadomi rodzinnego występowania choroby, jednak dopiero od 1946 roku zaczęto w sposób naukowy wyjaśniać podłoże genetyczne choroby. Kolejne lata owocują lawinowym rozwojem wiedzy w zakresie genetyki CF. W 1990 roku Green i Olson opracowali strategie klonowania i mapowania dużych regionów ludzkiego DNA.Badania dotyczyły także regionu chromosomu 7, w którym zlokalizowano gen CF.
Chociaż wydawać by się mogło, że istota mukowiscydozy jest już prawie całkowicie wyjaśniona, to efektywność leczenia jest nadal niewystarczająca. Znajomość historii CF, choroby „krwi, potu i łez”, „choroby o wielu maskach” prowokuje do refleksji co przyniesie przyszłość.
Tekst na podstawia http://www.muko.smolarnia.org/


